Soil Sodicity chemistry physics and amelioration
Author: Neal Menzies, Mike Bell, Peter Kopittke School of Agriculture and Food Sciences, The University of Queensland | Date: 25 Feb 2015
Take home message
At a mechanistic level, the adverse effects of sodicity on plant growth are reasonably well understood. Unfortunately, differences in soil and plant characteristics, climate and agronomy mean that this understanding cannot be directly converted to a simple set of fool-proof rules.
Like most cropping problems, growers and agronomists need to complement their knowledge of the underlying bio-physical system with careful observation to craft a solution appropriate for their situation.
Introduction
Sodicity, simply defined, is the presence of too much sodium (Na) in the soil. Unfortunately the next step of stating how much Na is too much, is not something we can so easily deal with. Part of this difficulty arises because of the usual differences we would find between different soils (differences in clay content, organic matter content, mineralogy, etc), and part arises because of the range of effects Na has on soils and on plant growth. The most commonly considered sodicity problem is decreased soil structural stability, and the resultant soil physical problems. We understand this problem, and have a number of amelioration strategies with which to address it. We less frequently consider how we should address the problem of sodicity resulting in excessively high pH (alkalinity). This problem is also well understood, and amelioration strategies are available, but in the Australian dry-land farming context their implementation is seldom economically attractive. Sodicity also results in a range of adverse impacts on plant nutrition, but this area is much less well understood. Indeed, it is the subject of considerable scientific debate, and I will provide you with an example of one of these science “conflicts” later in this paper.
This is an enormous subject, and it would take me a reasonable size book to relate the state of our understanding of the subject. For those of you who are interested, I refer you to the excellent book edited by Malcolm Sumner and Ravi Naidu “Sodic Soils, Distribution, Properties, Management and Environmental Consequences. 1998 Oxford University Press, New York. I have drawn on it liberally in this paper. Within this paper I will largely stick to the underlying science – the mechanisms by which Na creates problems in soils and by which ameliorants address these problems.
Effects of sodium on the physical properties of soil
Sodic soils have extremely poor physical characteristics which in agricultural soils lead to problems managing water and air regimes in the soil. The lack of soil structural stability results in dispersion of the surface during rainfall to form a seal. This seal limits infiltration partitioning a greater proportion of rainfall to runoff, and hence reduces water availability for crops growing in the soil, and increases the risk of erosion. On drying, the seal hardens as a crust which can prevent emergence of germinating seeds resulting in poor crop establishment. In addition, sodic soils are difficult to cultivate and have poor load-bearing characteristics. These behaviours are a result of the influence of Na on the clay fraction in the soil. When the cation exchange is occupied by Ca or Mg, the individual clay platelets aggregate as illustrated in Plate 1, and the soil behaves in many respects like a silt or sand because the aggregates of hundreds of clay platelets are of this physical size. The aggregated clay soil has good structural characteristics. When the exchange is occupied by Na, the individual clay platelets repel each other and these aggregates of clay platelets break up. The soil structure is destroyed, the clay disperses in water and is easily eroded. The breakaway gullies we commonly encounter in duplex soils are an excellent illustration of how easily Na saturated clays are eroded – the soil literally melts away. In a cultivated soil, much lower levels of Na saturation result in the various adverse agronomic outcomes as a result of only a small portion of the clay dispersing. It is always important to remember that sodicity is a problem that impacts on the clay fraction of the soil. In a sand with little clay fraction, sodicity will not result in adverse physical conditions (though there may still be adverse chemical effects as we will discuss later.

Plate 1. Scanning electron micrograph of aggregated clay platelets.

Plate 2. Breakaway gully formation illustrates the ease with which Na saturated clay can be dispersed and eroded.
The composition of the CEC dictates the soils’ physical behaviour, with dispersion being a complex issue dictated by exchange cation composition, clay mineralogy, soil texture, organic matter content, etc. Hence, rigid ‘rules’ and classification systems are not wonderful at predicting soil structural behaviour: this is where an understanding of the underlying processes (and a great deal of experience) are needed to permit us to manage soil sodicity.
At a mechanistic level, two processes, swelling and dispersion, are responsible for the behaviour of sodic soils, with these two processes governed by the soil surface charge and how it is balanced by exchangeable cations. So let’s start with a quick refresher on cation exchange capacity (CEC). Clay surfaces in most surface soils carry a net negative charge. This charge results in the cations being attracted to the surface, and these attracted cations balance the negative charge on the soil. Our starting conception of this arrangement is negative charge on the mineral, and a set of cations held tightly against the surface – we often see diagrams representing the system in this way (Figure 1a). This is wrong! The exchangeable cations are not held tightly against the surface, rather there is a higher concentration of cations in the soil solution near the surface where the cations have been attracted by the negative surface charge. Conversely, there are fewer anions in the soil solution near the surface, because they have been repelled by the negative surface charge. The zone of increased cation concentration and decreased anion concentration is called the diffuse double layer. Within this layer the sum of excess cations and deficit of anions equals the charge on the surface. So the surface charge is balanced not at the immediate surface, but within a distance from the surface (Figure 1b, Figure 2).
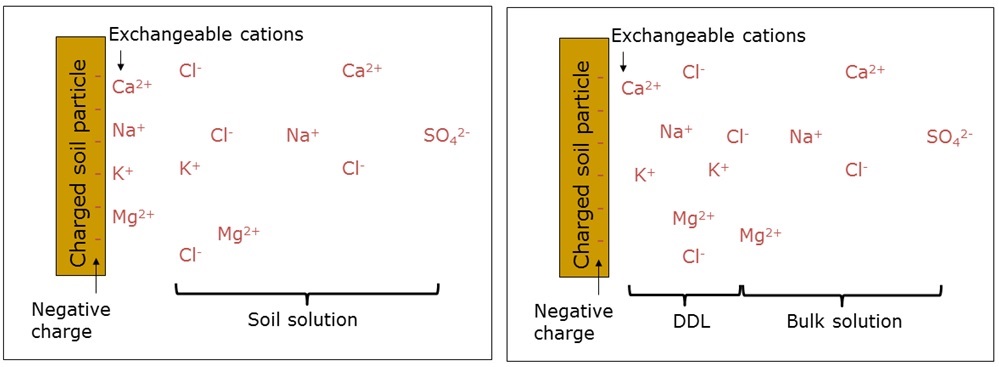
Figure 1. Schematic representation of the soil surface charge (cation exchange capacity) being balanced by (a) cations held tightly against the soil surface (an incorrect representation), and (b) by an excess of cations and deficit of anions in a volume of solution extending some distance from the surface (a correct representation).
Represented graphically (Figure 2), it is clear that the charge is largely balanced by cation excess (rather than anion deficit), and that distribution of cation excess is effectively an exponential decrease with distance from the surface.
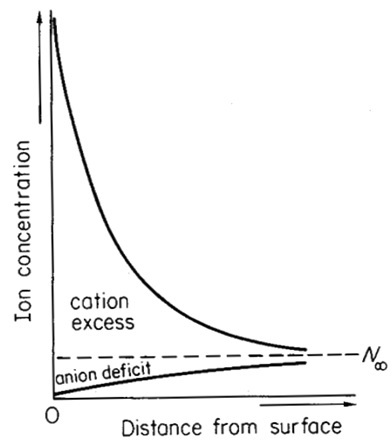
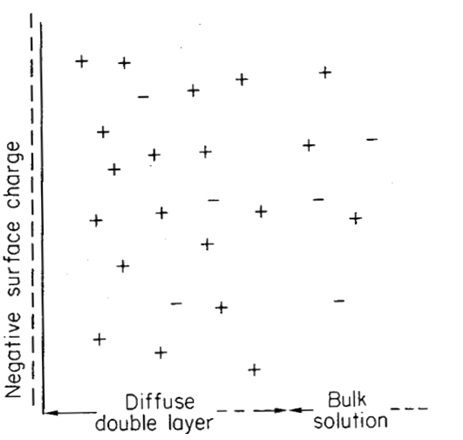
Figure 2. A representation of (lower) the distribution of cation and anions in the soil solution (analogous to Figure 1b), and (upper) a graph of the concentration of anions and cations with distance from the soil surface. In the diffuse double layer there is a higher concentration of cations than anions, while in the bulk solution the concentration of anions and cations is equal (represented by the dotted line). (This and following images are from van Olphen 1977 – I do not suggest that you read this, I am just acknowledging where I copied the figures from).
If we could design an appropriate probe that could sense charge that was not balanced by extra cations or fewer anions and gradually move this probe toward the soil surface, it would show no charge in the bulk soil solution, but show an exponentially increasing charge as we move it through the diffuse double layer toward the surface. Our imagined probe is measuring potential – the potential to do work.
If two charged soil particles approach each other until their diffuse double layers overlay, the work the potential can do is to push the soil particles away from each other. This is not the only force acting on the particle. Van der Waals forces attract particles to each other. The van der Waals forces are strong, but only act over a small distance. The balance between repulsion resulting from the surface charge and attraction resulting from van der Waals forces will determine if the particles come together (flocculate) or remain dispersed. The extent of the diffuse double layer can be changed, so we have the capacity to alter the balance of these two forces.
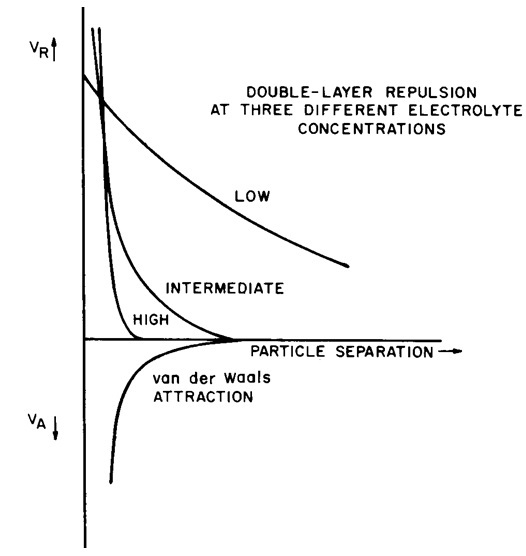
Figure 3. Representation of the strength (y axis) and distance of influence from the soil surface (x axis) of the van der Waals forces which attract particles together (force of attraction VA) and double layer repulsion (force of repulsion VR). The force of repulsion is plotted for three different soil solution concentrations (the effect of concentration on diffuse double layer thickness will be discussed shortly) The van der Waals forces are not affected by environmental influences, hence only one line is needed to represent these. (Figure copied from van Olphen 1977).
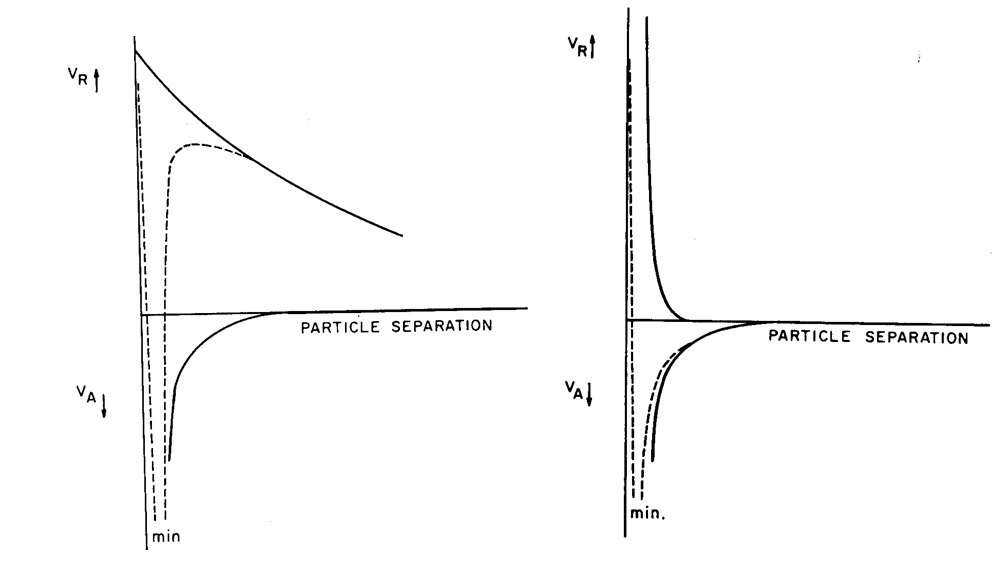
Figure 4. The attractive (below x axis) and repulsive (above x axis) forces acting on soil particles, and the net effect (dotted line) under conditions of (left) low ionic strength and (right) high ionic strength. (Figure from van Olphen 1977).
In low ionic strength (Figure 4 left) solutions where the diffuse double layer is broad, the counter ion atmospheres interact and create repulsive forces at much greater distances than the short range van der Waals attractive forces. Thus, flocculation of the soil is inhibited by an energy barrier. In the diagram below, the net energy balance (attraction/repulsion) is represented by the dotted line. This shows an initial high repulsion – the energy barrier to flocculation. There is then an energy-well; once the particles come close enough together, then they will attract each other. Given the large energy barrier, a system with this energy balance would take a long time to flocculate.
Note that there is a strong repulsion once the particles come close enough together – the cause of this is not represented by a solid line in the figure (so the dotted line is not a true sum of the two solid lines). This very short range repulsion is a result of two factors, Born repulsion (when extruding lattice points come into contact or interact), and the result of strong adsorption forces between the surface and water (normally surfaces are covered with one or two monomolecular layers of water).
Where the ionic strength of the soil solution is high (Figure 4 right) the repulsion force is at a minimum. Under this condition there is no energy barrier to flocculation. As two particles approach each other (because of their random motion in water), they will reach a point at which they are attracted, and they will flocculate. Such a system would rapidly flocculate.
Increasing the ionic strength of the electrolyte, or increasing the valance of the saturating cation (for example by replacing Na+ with Ca2+) causes compression of the counterion atmosphere and reduces the range over which the repulsive force acts. The attractive force is independent of concentration and valence of the counter ion. Maximum flocculation is induced at high concentration and high counter ion valance, when repulsive energy decays rapidly and attractive forces dominate at all distances from the surface (close in short range repulsive forces remain dominant).
The most common ameliorant applied to sodic soils to correct soil structural problems is gypsum. It acts to promote flocculation through both mechanisms; increasing soil solution ionic strength and by supplying divalent Ca ions to displace Na from exchange sites. The first of these effects (increased ionic strength) is immediate and can be achieved by relatively low rates of gypsum application, but the effect is short lived (especially if the application rate is low). Gypsum application can be particularly effective as a means of improving soil surface conditions at sowing, providing better soil tilth and reducing crusting. Clearly timing is important to ensure that rainfall and/or irrigation do not dissolve and leach all of the gypsum before sowing.
The effect of changing the counterion valance (replacing Na with Ca) is permanent (unless additional Na is supplied which will displace the Ca; for example through the use of poor quality irrigation water). Rates of gypsum application to displace Na from the exchange can be substantial. In Table 2, I have provided data for a Grey Clay soil and the calculated rate of gypsum required to reduce the Na saturation of the CEC to below 5%. The assumptions here are that replacement is perfect, i.e. that all of the Ca from gypsum replaces Na and none is leached, the gypsum is pure, and the soil bulk density is 1.2g/cm3. For this soil, 17 t/ha would be required to replace Na to 60 cm, and 33 t to treat 90 cm. Clearly these rates of application would not be economically attractive! Even if they were, we would need to consider the time it would take to move the gypsum derived Ca through the soil profile. Gypsum solubility is approximately 2.5 g/L, so it will take 40 mm of rainfall to dissolve 1 t/ha of gypsum. In an environment with 700 mm/year rainfall, it would take 2 years for the gypsum to dissolve from the soil surface. However it will take a great deal longer for the gypsum to move to depth. If we assume that 10 mm/year leaches beyond 90 cm (this estimate is derived from salinity / leaching work on the western Darling Downs), then it would take 65 years for the gypsum to reach the bottom of the soil profile.
Table 1. Exchangeable cation data for a grey clay soil, and the estimate gypsum requirement for different depth increments in the profile, and rainfall required to leach this gypsum into the soil.
|
Depth |
CEC |
Na |
5% of CEC |
Na reduction |
Gypsum |
Leaching |
|
cm |
cmol(+)/kg |
cmol(+)/kg |
cmol(+)/kg |
cmol(+)/kg |
t/ha |
mm |
|
0-10 |
29 |
2.3 |
1.5 |
0.8 |
0.8 |
30 |
|
10-20 |
30 |
4.2 |
1.5 |
2.7 |
2.8 |
110 |
|
20-30 |
29 |
4.0 |
1.5 |
2.5 |
2.6 |
100 |
|
30-60 |
32 |
5.1 |
1.6 |
3.5 |
10.8 |
430 |
|
60-90 |
30 |
6.8 |
1.5 |
5.3 |
16.3 |
650 |
|
Total |
33.3 |
1320 |
||||
Generally gypsum is applied at much lower rates than are required to displace all of the Na. The expectation from these smaller additions is that they will help to ameliorate the surface soil, increasing infiltration, and encouraging more uniform crop establishment. Repeat applications may be needed to sustain the surface soil improvement, and would certainly be needed if an impact on the subsoil sodicity was sought. Such small applications can be economically attractive. In the GRDC funded Combating Subsoil Constraints project (SIP08) one-time surface applied gypsum @2.5/ha increased cumulative gross margins by $207/ha over 4 crops (wheat 05, chickpea 07, wheat 08 and sorghum 09-10), reduced 115 t sodium chloride from the rooting depth and increased plant available water capacity by 15 mm (Dang et al 2010). Unfortunately, gypsum application is not always profitable, and more effective prediction of gypsum response is needed.
Note that the application of lime (CaCO3) also supplies Ca to the soil, but the low solubility of lime in neutral and alkaline soils prevents Ca release to participate in exchange reactions. Lime application to an acid sodic soil will address both the acidity and sodicity. Application of lime to a neutral or alkaline sodic soil will have little or no effect.
Some of the adverse soil structural aspects of sodicity may be addressed by increasing the organic matter content of soils – the organic matter acting to bind soil aggregates sustaining soil structure. There are a lot of reasons why growers would wish to increase the organic matter content of their soils; unfortunately, it is not an easy thing to achieve.
Effects of sodium on the chemical properties of soil
Cation nutrition
As the extent of Na saturation of the CEC increases, the reservoir of cationic plant nutrients (Ca, Mg, K) is diminished, and the ratio of Na to the other cations in soil solution increases dramatically. The selectivity of the soil CEC, with divalent ions held more strongly than monovalent cations, means that Na will be the dominant ion in soil solution, even when it only occupies a small proportion of the CEC. This is well demonstrated for a range of non-sodic soils in Figure 5. In these soils there is a trivial level of exchangeable Na, but Na is the dominant cation in soil solution. In saline soils, the Na dominance of the soil solution can be even more pronounced; in a saline vertisol with 11% of the CEC occupied by Na, but 81% occupied by Ca, the ratio of Na to Ca in the soil solution was more than 10:1 (100 mM Na, 8 mM Ca) (Dyer et al 2008).
The most important of the cation nutrition problems induced by sodicity is Ca deficiency, where high solution concentrations of Na interfere with plant uptake of Ca. It has long been recognised that Na is not the only cation which has this effect (high Mg readily induces Ca deficiency), and that the ratio of Ca to the total cations in solution is a better predictor of Ca deficiency than Ca concentration alone. An even more accurate prediction of Ca deficiency is obtained when expressed as a ratio of activity in solution, the calcium activity ratio (CAR) (see Figure 7).
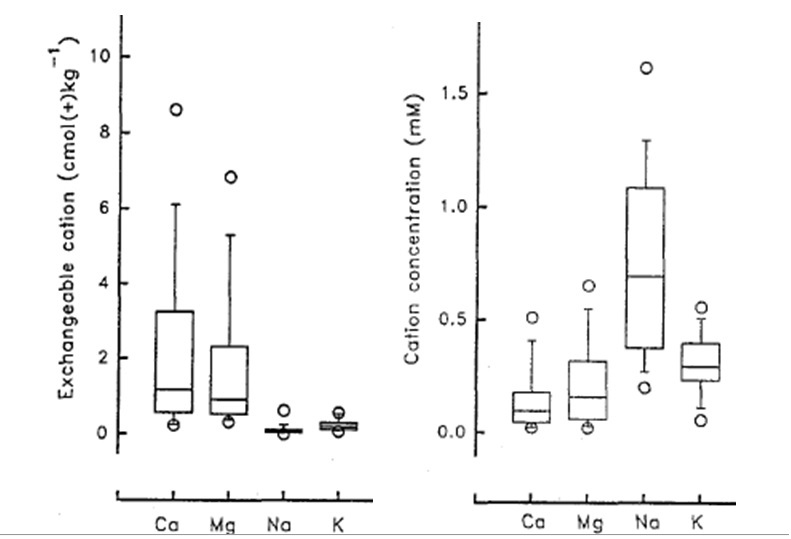
Figure 6. The distribution of exchangeable cations (left) and soil solution cation concentration in 60 non-saline / non-sodic surface soils. In each box plot the central line represents the median, the box captures 50% of the data points (From Menzies et al 1994)
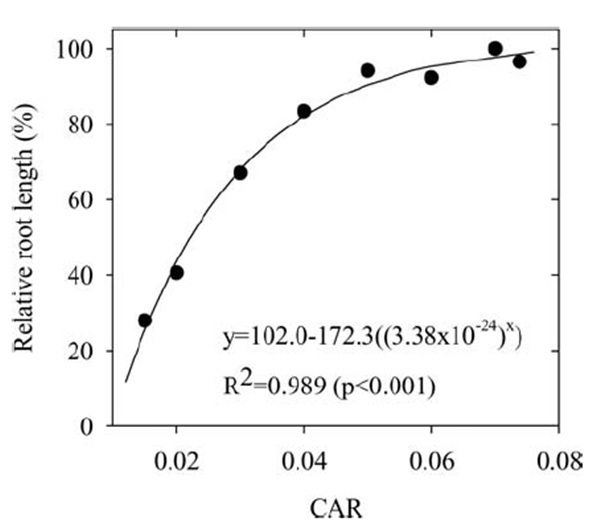
Figure 7. Relative root length of mungbean in nutrient solutions containing a constant concentration of Ca (0.60 mM) and Na (5 mM), where the Ca activity ratio (CAR) was manipulated by the addition of Mg. (From Kopittke and Menzies 2005)
Calcium has an important role in stabilizing the pectins in plant cell walls. Calcium cannot be readily translocated within the plant, so for roots to grow into soil, there must be sufficient Ca available in the soil solution within that soil volume. Thus Ca deficiency has a direct impact on root growth (Plate 3), with the resultant poor root system indirectly impacting the plant through the inability of the restricted root system to acquire water and nutrients. A crop growing in a soil where sodicity induced Ca deficiency at depth has limited root proliferation into the subsoil will be more susceptible to drought and less able to obtain nutrients at depth, rather than showing symptoms of Ca deficiency on the shoots.
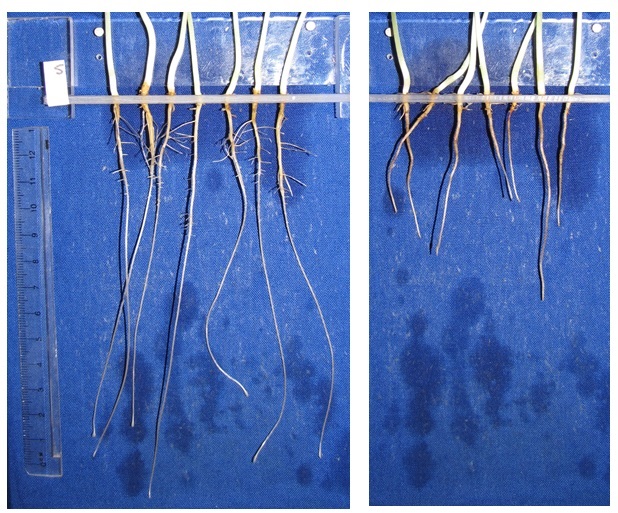
Plate 3. Cowpea root growth where adequately supplied with calcium (left) and plants grown at the same Ca concentration, but with sufficient Mg to induce Ca deficiency. Note that the Ca deficient roots are not only much shorter, but also that lateral root growth is severely impacted. Although not apparent in this plate, Ca deficient plants cannot produce healthy root hairs, and this will profoundly impact the plants’ capacity to capture nutrients like phosphorus.
Direct Na toxicity
Most plants are not especially sensitive to Na, with yield primarily being reduced by the osmotic effect of elevated Na concentration (salinity) rather than a direct toxicity of Na. Nevertheless, a portion of the yield reduction can be demonstrated to be the result of a specific Na toxicity. In an effort to tease out the separate effects of the osmotic stress and Na toxicity, Sheldon (2005) compared the growth of wheat when exposed to increasing concentrations of NaCl and when exposed to solutions of similar osmotic potential composed of a mixture of chloride salts (NaCl, KCl, MgCl2, CaCl2). The extent of Na toxicity can be assessed by a comparison of the yield of plants exposed to the same osmotic potential achieved using a mixture of cations to that achieved using Na alone (Figure 8). The lower yield for the Na only plants is a result of Na toxicity. In contrast, an analogous experiment comparing solutions where the anion component is Cl alone or a mixture of anions, shows that Cl is not toxic to wheat. Thus wheat growing in a saline (NaCl) soil is limited by salinity and Na toxicity, but not Cl toxicity.
Plant tolerance of Na varies extremely widely, with two broadly defined groups recognised: the glycophytes which are salt-sensitive, and the salt-tolerant halophytes. Most cultivated crops are glycophytes. Glycophytes rely on limiting uptake of Na, but because the plant uses a charge differential between the soil solution and the electronegative interior of the cell to drive the uptake of cationic nutrients, and because the cation transporters in the plasma membrane are to some extent permeable to Na, it is not possible for the plant to prevent Na accumulation. In contrast, halophytes do not restrict Na uptake, but compartmentalize the Na into cellular vacuoles as a useful osmotic regulation strategy to assist in dealing with saline soils. The halophyte strategy permits the plant to survive in saline environments, and there may be merit in considering this strategy in breeding salt-tolerant crops – but it is worth remembering that this is a “survival” strategy; halophytes are not known for their rapid growth / high yield!
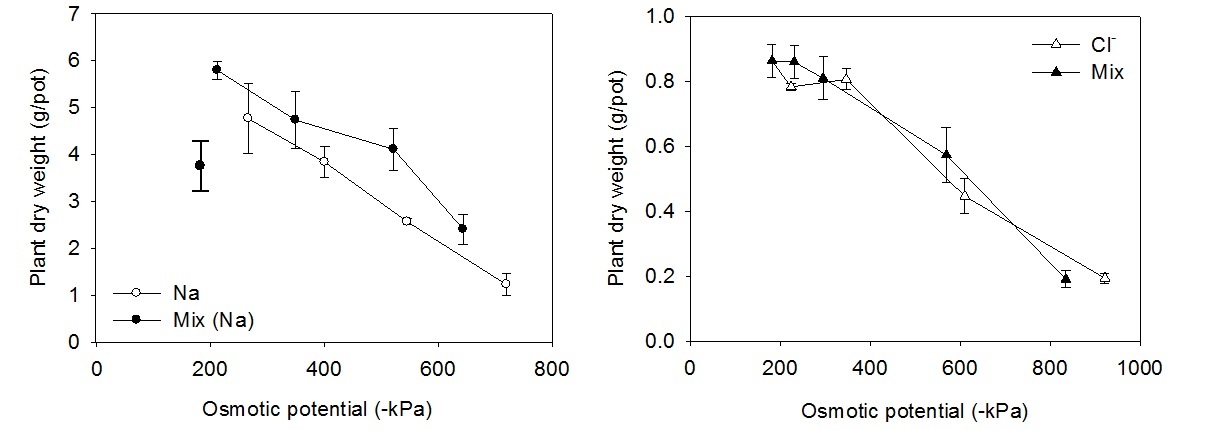
Figure 8. The dry-weight of shoots compared to the osmotic potential of the treatment solution. In the left hand figure the open symbols represent treatments where all the cations were Na+, in the right hand figure they represent treatments where all of the anions are Cl-. The solid symbols indicate treatments that contained mixed anions and cations (From Shelton et al 2006).
Alkalinity (and the vexed issue of aluminate toxicity)
Where Na is the dominant cation in soil solution, the pH can rise to higher values than occurs where Ca or Mg is the dominant cation. This is a simple reflection of the solubility of the respective carbonates of these cations. Calcium and Mg carbonates are not very soluble, while Na and K carbonates dissolve readily in water to form solutions with very high pH (Table 2), according to the reactions;
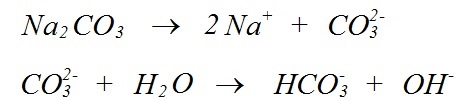
Table 2. The solubility of carbonate salts in water.
More soluble salts will support a higher solution pH.
|
Carbonate |
Solubility (g/L) |
|
CaCO3 |
0.014 |
|
MgCO3 |
1.76 |
|
Na2CO3 |
71 |
|
K2CO3 |
1120 |
The alkalinity of Ca2+-rich soils is precipitated in the form of CaCO3 (calcite), which controls the solution pH at a value near 8.2 (assuming a CO2 gas pressure of 0.3 millibar). These soils are termed calcareous, and are not considered to be alkaline despite their pH, because they do not possess the negative attributes of the more strongly alkaline soils. If on the other hand, alkalinity is present in the form of Na (or K) bicarbonates and carbonates, the soil can have a very high pH and pose an alkalinity hazard to plants.
This issue of solubility of carbonates provides us with the mechanism by which the most widely used ameliorant for alkaline soils, gypsum, acts. Application of gypsum to the soil supplies Ca ions which precipitate as CaCO3 consuming alkalinity. Effectively the reverse of the equations written above for sodium carbonate. Each mole of gypsum applied has the capacity to precipitate 2 moles of alkalinity. This alkalinity is not removed from the soil, it remains present in the precipitated carbonate, with the pH lowered to the 8.2 to 8.5 range. Another commonly used ameliorant for alkaline soils is elemental sulfur, which is oxidised by bacteria in the soil, effectively producing sulfuric acid. In this instance the alkalinity is actually removed from the soil. One of the limitations of this approach is the need to predict the elemental S application accurately to prevent an excessive application producing an acid soil. In contrast, an excessive application of gypsum will not harm the soil (it is simply a waste of money).
Plant growth at high pH is limited indirectly both by nutritional disorders and HCO- toxicity. Iron deficiency (lime-induced chlorosis) is the most common nutritional disorder of plants growing on alkaline soils and is caused both by low Fe availability and by elevated concentrations of HCO-. The availability of other nutrients such as Ca, Zn, P and Mn, is also low in alkaline conditions due mainly to adsorption and precipitation processes. Alkalinity itself (i.e. the OH- ion) is also directly detrimental to plant roots, but its' influence is typically overshadowed by the problems induced by high pH. Very careful experimentation is required in order to investigate the effect of high pH per se, but when sufficient care is taken, OH- can be shown to reduce plant growth at pH values commonly encountered in soils (e.g. mungbean root length was decreased at pH 8.5 Kopittke and Menzies 2004). We will return to this need for careful experimentation shortly when we consider aluminium (Al) toxicity at high pH.
In acid soils the toxicity of Al3+ and other monomeric hydroxy-Al species (AlOH2+, Al(OH)2+) at low concentration is well established. The critical Al concentration at which root elongation is inhibited is as low as 5 to 20 mM in solutions of low ionic strength representative of acid soils. To ameliorate acid, Al-toxic soils, lime is applied to raise the soil pH, as Al solubility decreases with increasing pH. Where we encounter acid sodic soils, lime acts as an ameliorant for the detrimental effects of both the acidity and the sodicity by raising the pH and supplying Ca ions into the soil solution.
Interestingly, the solubility of Al increases again at high pH (Figure 9), with the Al in an anionic form as aluminate (Al(OH)4-). At a mechanistic level, it does not seem sensible to assume that Al in an anionic form will be toxic in the same manner as it is as a cation, and much of the evidence used to establish aluminate toxicity is based on correlation, rather than a demonstration of causation. By this we mean that measurements of Al in soil solution show an increase as soil pH increases in alkaline soils, and at the same time, plant growth decreases as pH increases – hence the correlation between increasing aluminate and decreasing plant growth, and the basis of the claim that aluminate is toxic to plants. Of course this correlation ignores the rapidly decreasing availability of Ca and Fe, and the increasing toxicity of HCO3-, etc. So correlation evidence is little better than no evidence at all!
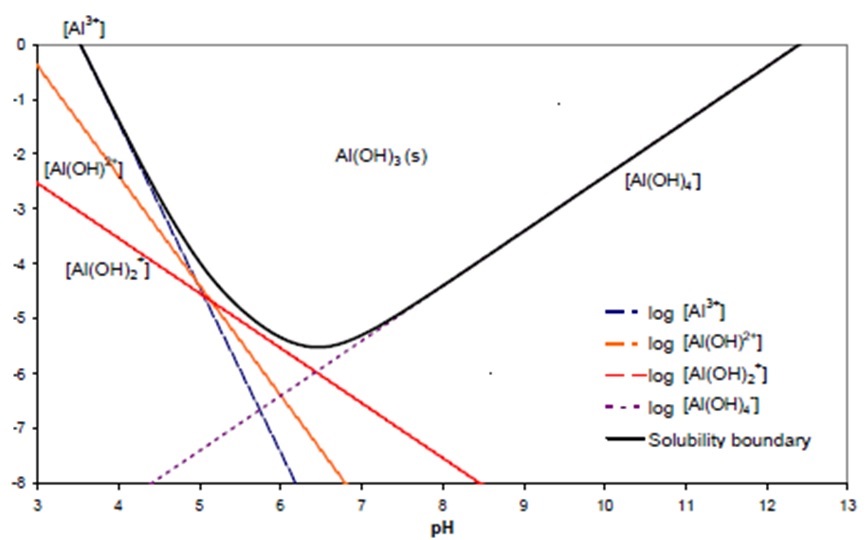
Figure 9. The concentration of various forms of aluminium in solution in equilibrium with aluminium hydroxide at a range of pH values. The solid black line represents the total concentration in solution. Note that the y-axis units are Log (molar concentration)
Other researchers have taken a more direct approach to demonstrating aluminate toxicity by comparing plant growth in alkaline solutions which are aluminate free (e.g. Na2CO3 solution), to growth in the same solution to which Al has been added to test the toxicity of aluminate. Plants grow less well in the Al containing solution, and this is interpreted a clear evidence (causation evidence). Unfortunately, it’s not that simple! When Al is added to an alkaline solution, some of the added Al forms an Al13 polymer (Figure 10), and this is profoundly toxic with only 3 to 4 µM sufficient to fully inhibit wheat root growth (Parker et al 1989). Of course, if you do not look for polymeric Al, you will not find it, and you will be happy to say that aluminate is toxic. We went to extraordinary lengths to prevent the formation of polymeric Al in order to establish if aluminate was toxic, yet still found Al13 forming over time in our solution culture systems so that we could only establish that aluminate was not toxic by regular replacement of the test solutions (Kopittke et al 2004). An important additional consideration then is whether Al13 will be a limitation to plants growing in alkaline soils. This seems very unlikely. It will not form where silicate or sulfate is present, it is thermodynamically unstable and will eventually precipitate as Al(OH)3 with this process speeded by the presence of soil minerals, and finally it has a charge of 7+ and so would be held extremely strongly by the soil CEC.
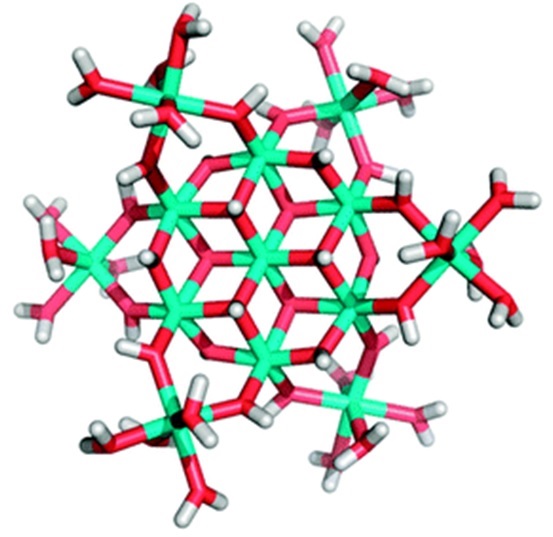
Figure 10. A stick model of the Al13 polymer. Al atoms are represented by the green intersection points, with oxygen represented by red and hydrogen by white. This polymer is profoundly toxic to plants, but is only likely to be encountered as an experimental artefact in solution culture experiments. Its' high surface charge of 7 would see it held very strongly by the CEC in soil.
Some concluding comments
While we have discussed the various physical and chemical effects of sodicity (alkalinity) in isolation, the reality is that they will occur simultaneously in a sodic soil. Furthermore, the observable impact of the various adverse soil conditions on plant roots will be similar, so it is very difficult to attribute the plant growth limitation to a particular mechanism. For example poor soil structure will result in susceptibility to waterlogging, with the roots damaged (killed) by low oxygen availability, but these damaged roots would not be readily distinguished from roots damaged by Ca deficiency or by alkalinity. At a whole plant level each of these problems (or the combination of all of these problems) will result in drought susceptibility, poor capacity to capture nutrients like phosphorus which are obtained by active uptake and diffusion toward the root.
In the majority of instances, we do not need to know the precise mechanistic nature of the problem, as the same ameliorative strategy, the application of soluble Ca (most commonly as gypsum), will address most of the limitations. Nevertheless, some knowledge of the specific problem faced (often gained by thoughtful observation) will guide the implementation of a remediation strategy. For example, the various aspects of poor soil structure caused by dispersion are a diffuse double layer problem, but individual expressions of poor soil structure require different remediation strategies. At the immediate surface of the soil, dispersion can result in surface sealing, and in the short term this can be addressed by increasing the ionic strength of the soil solution through the application of relatively low rates of gypsum. These applications must be repeated regularly as rainfall will dissolve the gypsum and leach it down through the soil profile, once the solid phase gypsum is all dissolved, the ionic strength of the soil solution will fall – approaching the very low ionic strength of rainwater at the soil surface – and the risk of surface sealing will re-emerge. Deeper in the soil profile, the ionic strength of the soil solution is much more buffered, and the beneficial effect of gypsum application is limited to the replacement of Na by Ca on the CEC.
Numerous important knowledge gaps remain in our understanding of sodic soils. Stating these in pragmatic terms (rather than as the underlying science), we need
- To be better able to predict on which soils an economic benefit will be gained from the application of gypsum (including setting the appropriate rate of application, and frequency of repeat applications)
- To develop strategies for the amelioration of sodic subsoils, and the ability to predict when subsoil amelioration will be economically attractive
- To refine water and nutrient management approaches for sodic soils.
- To understand, and hence be able to optimize, alternative amelioration strategies (e.g. organic matter management).
References
Dang YP, Dalal RC, Buck SR, Harms B, Kelly R, Hochman Z, Schwenke GD, Biggs AJW, Ferguson N, Norrish S, Routley R, McDonald M, Hall C, Singh J, Daniells IJ, Farquharson R, Manning W, Speirs S, Grewal HS, Cornish P, Bodapati N, Orange D (2010) Diagnosis, extent, impacts and management of subsoil constraints in the northern grains cropping region of Australia. Australian Journal of Soil Research 48, 105–119.
Dyer CL, Kopittke PM, Sheldon AR, Menzies NW (2008) Influence of soil moisture content on soil solution composition. Soil Science Society of America Journal 72, 355-361.
Kopittke PM, Menzies NW (2004) Control of nutrient solutions for studies at high pH. Plant and Soil 266, 343-354.
Kopittke PM, Menzies NW, Blamey FPC (2004) Rhizotoxicity of aluminate and polycationic aluminum at high pH. Plant and Soil 266, 177-186.
Kopittke PM, Menzies NW (2005) Effect of pH on Na induced Ca deficiency. Plant and Soil 269, 119-129.
Menzies NW, Bell LC, Edwards DG (1994) Exchange and solution phase chemistry of acid, highly weathered soils. I. Characteristics of soils and the effects of lime and gypsum amendments. Australian Journal of Soil Research 32, 251-267.
Parker DR, Kinraide TB, Zelazny LW (1989) On the phytotoxicity of polynuclear hydroxy-aluminum complexes. Soil Science Society of America Journal 53, 789-796.
Sheldon AR, Dalal RC, Menzies NW Toxicity of Na+ and Cl- to wheat and chickpea. In '18th World Congress of Soil Science. International Union of Soil Science', 2006, Philadelphia, Pennsylvania, USA,
Sumner ME, Naidu R (1998) 'Sodic Soils: Distribution, Properties, Management, and Environmental Consequences.' (Oxford University Press: New York)
van Olphen H (1977) 'An Introduction to Clay Colloid Chemistry.' (Wiley - Interscience: New York)
Contact details
Neal Menzies
School of Agriculture and Food Sciences
The University of Queensland
Email: n.menzies@uq.edu.au
Was this page helpful?
YOUR FEEDBACK